| 生物活性 | Valproic acid (VPA) is an orally activeHDACinhibitor, withIC50in the range of 0.5 and 2 mM, also inhibitsHDAC1(IC50, 400 μM), and induces proteasomal degradation ofHDAC2. Valproic acid activatesNotch1signaling and inhibits proliferation in small cell lungcancer(SCLC) cells. Valproic acid is used in the treatment of epilepsy, bipolar disorder,metabolic disease, HIVinfectionand prevention of migraine headaches[1][2][3][4][5][6][7]. |
| IC50& Target[5][6] | HDAC1 400 μM (IC50) | HDAC 0.5-2 mM (IC50) | HDAC2 | Autophagy | Mitophagy |
|
体外研究
(In Vitro) | Valproic acid (VPA) (0-15 mM; 24 and 72 h) inhibits Hela cell growth in a dose- and time- dependent manner[1].
Valproic acid (10 mM; 24 h) significantly attenuates the activities of total, cytosol and nuclear HDACs[1].
Valproic acid (0-15 mM; 24 h) induces a G1 phase arrest at 1–3 mM and a G2/M phase arrest at 10 mM, and increases the percentage of sub-G1 cells in HeLa cells. Valproic acid also induces necrosis, apoptosis and lactate dehydrogenase (LDH) release[1].
Valproic acid (0-20 mM; 24 h) activates Tcf/Lef-dependent transcription and synergizes with lithium[2].
Valproic acid (0-5 mM; 0-18 h) increases β-catenin levels in Neuro2A cells[2].
Valproic acid (0-2 mM; 0-24 h) stimulates phosphorylation of AMPK and ACC in hepatocytes[5].
Valproic acid (0-10 mM; 2 days) induces Notch1 signaling and morphologic differentiation, suppresses production of NE tumor markers in SCLC cells[6].
Cell Viability Assay[1] | Cell Line: | HeLa cells | | Concentration: | 0, 1, 3, 5, 10 and 15 mM | | Incubation Time: | 24 and 72 h | | Result: | HeLa cell growth was dose- and time-dependently decreased with an IC50of ~10 and 4 mM at 24 and 72 h. |
Western Blot Analysis[1][2][5] | Cell Line: | HeLa cells, Neuro2A cells or primary mouse hepatocytes | | Concentration: | 10 mM (HeLa); 0, 2, and 5 mM (Neuro2A); 0.2, 0.4, 0.8, 1.2 and 2 mM (hepatocytes) | | Incubation Time: | 10 mM (HeLa); 0, 2, and 5 mM (Neuro2A); 0.2, 0.4, 0.8, 1.2 and 2 Mm (hepatocytes) | | Result: | Increased the form of acetylated histone 3.
Reduced PARP, induced cleavage PARP, and downregulated Bcl-2.
Increased β-catenin levels.
Increased the phosphorylation of AMPK and ACC. |
Cell Cycle Analysis[1] | Cell Line: | HeLa cells | | Concentration: | 0, 1, 3, 5, 10 and 15 mM | | Incubation Time: | 24 h | | Result: | Induced a G1 phase arrest at 1–3 mM, significantly induced a G2/M phase arrest at 10 mM, and increased the percentage of sub-G1 cells in HeLa cells in a dose-dependent manner at 24 h. |
|
体内研究
(In Vivo) | Valproic acid (VPA) (500 mg/kg; i.p.; daily for 12 days) inhibits tumor angiogenesis in mice transplanted with Kasumi-1 cells[3].
Valproic acid (350 mg/kg; i.p.; once) enhances social behavior in rats[4].
Valproic acid (0.26% (w/v); p.o. via drinking water; 14 days) decreases liver mass, hepatic fat accumulation, and serum glucose in obese mice without hepatotoxicity[5].
| Animal Model: | Female BALB/c nude mice, Kasumi-1 tumor model[3] | | Dosage: | 500 mg/kg | | Administration: | Intraperitoneal injection, daily for 12 days | | Result: | Inhibited tumor growth and tumor angiogenesis.
Inhibited the mRNA and protein expression of VEGF, VEGFR2 and bFGF.
Inhibited HDAC activity and increased acetylation of histone H3.
Enhanced the accumulation of hyperacetylated histone H3 on VEGF promoters. |
| Animal Model: | Timed-pregnant Long Evans rats[4] | | Dosage: | 350 mg/kg | | Administration: | Intraperitoneal injection, once | | Result: | Demonstrated more social investigation and play fighting than control animals. |
| Animal Model: | Obese phenotype of ob/ob mice[5] | | Dosage: | 0.26% (w/v) | | Administration: | Oral via drinking water, 14 days | | Result: | Revealed a marked reduction in the accumulation of fats in the liver as compared with the untreated mice, significantly decreased liver mass to body mass, decreased serum triglyceride concentrations, and did not induce hepatotoxicity. |
|
| 分子量 | |
| 性状 | |
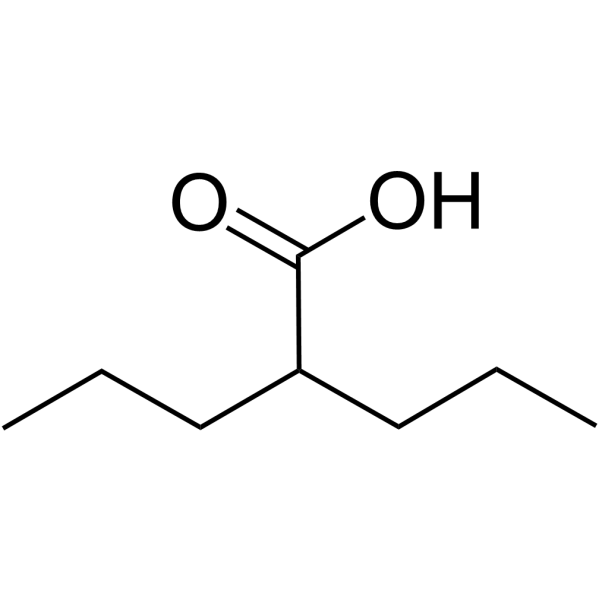
| Formula | |
| CAS 号 | |
| 中文名称 | |
| 运输条件 | Room temperature in continental US; may vary elsewhere. |
| 储存方式 | | Pure form | -20°C | 3 years | | 4°C | 2 years | | In solvent | -80°C | 6 months | | -20°C | 1 month |
|
| 溶解性数据 | In Vitro: DMSO : 100 mg/mL(693.43 mM;Need ultrasonic) H2O : 1 mg/mL(6.93 mM;Need ultrasonic and warming) 配制储备液 | 1 mM | 6.9343 mL | 34.6717 mL | 69.3433 mL | | 5 mM | 1.3869 mL | 6.9343 mL | 13.8687 mL | | 10 mM | 0.6934 mL | 3.4672 mL | 6.9343 mL |
*请根据产品在不同溶剂中的溶解度选择合适的溶剂配制储备液;一旦配成溶液,请分装保存,避免反复冻融造成的产品失效。
储备液的保存方式和期限:-80℃, 6 months; -20℃, 1 month。-80℃ 储存时,请在 6 个月内使用,-20℃ 储存时,请在 1 个月内使用。 In Vivo: 请根据您的实验动物和给药方式选择适当的溶解方案。以下溶解方案都请先按照In Vitro方式配制澄清的储备液,再依次添加助溶剂: ——为保证实验结果的可靠性,澄清的储备液可以根据储存条件,适当保存;体内实验的工作液,建议您现用现配,当天使用;
以下溶剂前显示的百
分比是指该溶剂在您配制终溶液中的体积占比;如在配制过程中出现沉淀、析出现象,可以通过加热和/或超声的方式助溶 1. 请依序添加每种溶剂: PBS Solubility: 100 mg/mL (693.43 mM); Clear solution; Need ultrasonic and warming and heat to 60℃ 2. 请依序添加每种溶剂: 0.5% CMC/saline water Solubility: 20 mg/mL (138.69 mM); Suspended solution; Need ultrasonic 3. 请依序添加每种溶剂: 10% DMSO 40%PEG300 5%Tween-80 45% saline Solubility: ≥ 2.5 mg/mL (17.34 mM); Clear solution
此方案可获得 ≥ 2.5 mg/mL (17.34 mM,饱和度未知) 的澄清溶液。 以 1 mL 工作液为例,取 100 μL 25.0 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀;向上述体系中加入50 μL Tween-80,混合均匀;然后继续加入 450 μL生理盐水定容至 1 mL。 4. 请依序添加每种溶剂: 10% DMSO 90% (20%SBE-β-CDin saline) Solubility: ≥ 2.5 mg/mL (17.34 mM); Clear solution
此方案可获得 ≥ 2.5 mg/mL (17.34 mM,饱和度未知) 的澄清溶液。 以 1 mL 工作液为例,取 100 μL 25.0 mg/mL 的澄清 DMSO 储备液加到 900 μL20% 的 SBE-β-CD 生理盐水水溶液中,混合均匀。 5. 请依序添加每种溶剂: 10% DMSO 90%corn oil Solubility: ≥ 2.5 mg/mL (17.34 mM); Clear solution
此方案可获得 ≥ 2.5 mg/mL (17.34 mM,饱和度未知) 的澄清溶液,此方案不适用于实验周期在半个月以上的实验。 以 1 mL 工作液为例,取 100 μL 25.0 mg/mL 的澄清 DMSO 储备液加到 900 μL玉米油中,混合均匀。 *以上所有助溶剂都可在本网站选购。
|
 m.cnreagent.com
m.cnreagent.com