| CAS NO: | 341031-54-7 |
| 规格: | ≥98% |
| 包装 | 价格(元) |
| 100mg | 电议 |
| 500mg | 电议 |
| 1g | 电议 |
| 5g | 电议 |
| 10g | 电议 |
| Molecular Weight (MW) | 532.56 |
|---|---|
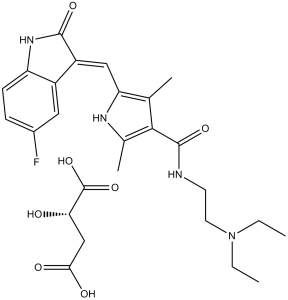
| Formula | C22H27FN4O2.C4H6O5 |
| CAS No. | 341031-54-7 (malate); |
| Storage | -20℃ for 3 years in powder form |
| -80℃ for 2 years in solvent | |
| Solubility (In vitro) | DMSO: 15 mg/mL (28.2 mM) |
| Water: <1 mg/mL | |
| Ethanol:<1 mg/mL | |
| Solubility (In vivo) | 4% DMSO+30% PEG 300+ddH2O: 2 mg/mL |
| Synonyms | sunitinib; SU-11248; SU 11248; SU11248; SU011248. trade name: Sutent. Chemical Name: (Z)-N-(2-(diethylamino)ethyl)-5-((5-fluoro-2-oxoindolin-3-ylidene)methyl)-2,4-dimethyl-1H-pyrrole-3-carboxamide malate InChi Key: LBWFXVZLPYTWQI-HBPAQXCTSA-N InChi Code: InChI=1S/C22H27FN4O2.C4H6O5/c1-5-27(6-2)10-9-24-22(29)20-13(3)19(25-14(20)4)12-17-16-11-15(23)7-8-18(16)26-21(17)28;5-2(4(8)9)1-3(6)7/h7-8,11-12,25H,5-6,9-10H2,1-4H3,(H,24,29)(H,26,28);2,5H,1H2,(H,6,7)(H,8,9)/b17-12-; SMILES Code: O=C(C1=C(C)NC(/C=C2C(NC3=C\2C=C(F)C=C3)=O)=C1C)NCCN(CC)CC.OC(CC(O)=O)C(O)=O |
| In Vitro | In vitro activity: Sunitinib also potently inhibits Kit and FLT-3. Sunitinib is a potent ATP-competitive inhibitor of VEGFR2 (Flk1) and PDGFRβ with Ki of 9 nM and 8 nM, respectively, displaying>10-fold higher selectivity for VEGFR2 and PDGFR than FGFR-1, EGFR, Cdk2, Met, IGFR-1, Abl, and src. In serum-starved NIH-3T3 cells expressing VEGFR2 or PDGFRβ, Sunitinib inhibits VEGF-dependent VEGFR2 phosphorylation and PDGF-dependent PDGFRβ phosphorylation with IC50 of 10 nM and 10 nM, respectively. Sunitinib inhibits VEGF-induced proliferation of serum-starved HUVECs with IC50 of 40 nM, and inhibits PDGF-induced proliferation of NIH-3T3 cells overexpressing PDGFRβ or PDGFRα with IC50 of 39 nM and 69 nM, respectively. Sunitinib inhibits phosphorylation of wild-type FLT3, FLT3-ITD, and FLT3-Asp835 with IC50 of 250 nM, 50 nM, and 30 nM, respectively. Sunitinib inhibits the proliferation of MV4;11 and OC1-AML5 cells with IC50 of 8 nM and 14 nM, respectively, and induces apoptosis in a dose-dependent manner. Kinase Assay: IC50 values for Sunitinib against VEGFR2 (Flk-1) and PDGFRβ are determined using glutathione S-transferasefusion proteins containing the complete cytoplasmic domain of the RTK. Biochemical tyrosine kinase assays to quantitate the trans-phosphorylation activity of VEGFR2 (Flk-1) and PDGFRβ are performed in 96-well microtiter plates precoated (20 μg/well in PBS; incubated overnight at 4 °C) with the peptide substrate poly-Glu,Tyr (4:1). Excess protein binding sites are blocked with the addition of 1-5% (w/v) BSA in PBS. Purified GST-fusion proteins are produced in baculovirus-infected insect cells. GST-VEGFR2 and GST-PDGFRβ are then added to the microtiter wells in 2 × concentration kinase dilution buffer consisting of 100 mM HEPES, 50 mM NaCl, 40 μM NaVO4, and 0.02% (w/v) BSA. The final enzyme concentration for GST-VEGFR2 or GST-PDGFRβ is 50 ng/mL. Twenty-five μL of diluted Sunitinib are subsequently added to each reaction well to produce a range of inhibitor concentrations appropriate for each enzyme. The kinase reaction is initiated by the addition of different concentrations of ATP in a solution of MnCl2 so that the final ATP concentrations spanned the Km for the enzyme, and the final concentration of MnCl2 is 10 mM. The plates are incubated for 5-15 minutes at room temperature before stopping the reaction with the addition of EDTA. The plates are then washed three times with TBST. Rabbit polyclonal antiphosphotyrosine antisera are added to the wells at a 1:10,000 dilution in TBST containing 0.5% (w/v) BSA, 0.025% (w/v) nonfat dry milk, and 100 μM NaVO4 and incubated for 1 hour at 37 °C. The plates are then washed three times with TBST, followed by the addition of goat antirabbit antisera conjugated with horseradish peroxidase (1:10,000 dilution in TBST). The plates are incubated for 1 hour at 37 °C and then washed three times with TBST. The amount of phosphotyrosine in each well is quantitated after the addition of 2,2′-azino-di-[3-ethylbenzthiazoline sulfonate] as substrate. Cell Assay: Cells are starved overnight in medium containing 0.1% FBS prior to addition of Sunitinib and FL (50 ng/mL; FLT3-WT cells only). Proliferation is measured after 48 hours of culture using the Alamar Blue assay or trypan blue cell viability assays. Apoptosis is measured 24 hours after Sunitinib addition by Western blotting to detect cleavage of poly (ADP-ribose) polymerase (PARP) or levels of caspase-3. |
|---|---|
| In Vivo | Consistent with the substantial and selective inhibition of VEGFR2 or PDGFR phosphorylation and signaling in vivo, Sunitinib (20-80 mg/kg/day) exhibits broad and potent dose-dependent anti-tumor activity against a variety of tumor xenograft models including HT-29, A431, Colo205, H-460, SF763T, C6, A375, or MDA-MB-435. Sunitinib dosing at 80 mg/kg/day for 21 days leads to complete tumor regression in six of eight mice, without tumor re-growing during a 110-day observation period after the end of treatment. Second round of treatment with Sunitinib remains efficacious against tumors that are not fully regressed during the first round of treatment. Sunitinib treatment results in significant decrease in tumor MVD, with ~40% reduction in SF763T glioma tumors. SU11248 treatment results in a complete inhibition of additional tumor growth of luciferase-expressing PC-3M xenografts, despite no reduction in tumor size. Sunitinib treatment (20 mg/kg/day) dramatically suppresses the growth subcutaneous MV4;11 (FLT3-ITD) xenografts and prolongs survival in the FLT3-ITD bone marrow engraftment model. |
| Animal model | Female nu/nu mice implanted s.c. with HT-29, A431, Colo205, H-460, SF763T, C6, A375, or MDA-MB-435, and male nu/nu mice bearing luciferase-expressing PC-3M tumors |
| Formulation & Dosage | Dissolved in carboxymethyl cellulose suspension or as a citrate buffered (pH 3.5) solution; 80mg/kg; Oral gavage |
| References | J Med Chem. 2003 Mar 27;46(7):1116-9; Clin Cancer Res. 2003 Jan;9(1):327-37; Blood. 2003 May 1;101(9):3597-605. |
 m.cnreagent.com
m.cnreagent.com