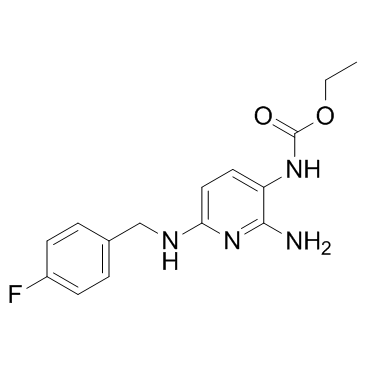
| CAS NO: | 56995-20-1 |
| 规格: | 98% |
| 分子量: | 304.32 |
| 包装 | 价格(元) |
| 10mg | 电议 |
| 50mg | 电议 |
| 100mg | 电议 |
| 500mg | 电议 |
Background:
Flupirtine(D 9998) is a selective neuronal potassium channel opener that also has NMDA receptor antagonist properties.IC50 Value: Target: Potassium channel; NMDA receptorin vitro: High concentrations of flupirtine antagonized inward currents to NMDA(200 microM) at -70 mV with an lC50 against steady-state responses of 182.1+/-12.1 microM. The effects of flupirtine were voltage-independent and not associated with receptor desensitization making actions within the NMDA receptor channel or at the glycine modulatory site unlikely. NMDA receptor antagonism probably has little relevance for the clinical efficacy of flupirtine as the concentrations needed were far higher than those achieved in clinical practice. However, the activation of a G-protein-regulated inwardly rectifying K+ channel was identified as an interesting molecular target site of flupirtine. In the next stage, the central nervous spectrum of action of experimental K+ channel openers (PCO) was considered. As far as they have been studied, experimental K+ channel openers display a spectrum of action comparable to that of flupirtine [1]. Therapeutic flupirtine concentrations (≤10 •M) did not affect voltage-gated Na(+) or Ca(2+) channels, inward rectifier K(+) channels, nicotinic acetylcholine receptors, glycine or ionotropic glutamate receptors. Flupirtine shifted the gating of K(V)7 K(+) channels to more negative potentials and the gating of GABA(A) receptors to lower GABA concentrations [2]. Cell exposure to flupirtine decreased the amplitude of delayed rectifier K(+) current (I(K(DR))) with a concomitant raise in current inactivation in NSC-34 neuronal cells [4].in vivo: Rats were trained to discriminate the novel analgesic flupirtine (10.0 mg/kg i.p., 10 min) from no drug under a two-choice fixed-ratio 5 shock-termination schedule. Flupirtine yielded a dose-response curve with an ED50 of 3.87 mg/kg. The opioid analgesics pentazocine, codeine and tramadol failed to produce flupirtine appropriate responding. The opioid antagonist naltrexone did not antagonize the discriminative effects of flupirtine [3]. Both morphine (ED•• =•0.74•mg/kg) and flupirtine (ED•••=•3.32•mg/kg) caused dose-related anti-hyperalgesia at doses that did not cause sedation [5]. Toxicity: Based on study-end data, hepatotoxicity was detected in 31% of patients receiving flupirtine for ≥ 6 weeks [6].
[1]. Kornhuber J, et al. Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltageindependent potassium channels. Rapid communication. J Neural Transm. 1999;106(9-10):857-67. [2]. Klinger F, et al. Concomitant facilitation of GABAA receptors and KV7 channels by the non-opioid analgesic flupirtine. Br J Pharmacol. 2012 Jul;166(5):1631-42. [3]. Swedberg MD, et al. Pharmacological mechanisms of action of flupirtine: a novel, centrally acting, nonopioid analgesic evaluated by its discriminative effects in the rat. J Pharmacol Exp Ther. 1988 Sep;246(3):1067-74. [4]. Wu SN, et al. Evidence for inhibitory effects of flupirtine, a centrally acting analgesic, on delayed rectifier k(+) currents in motor neuron-like cells. Evid Based Complement Alternat Med. 2012;2012:148403. [5]. Kolosov A, et al. Flupirtine enhances the anti-hyperalgesic effects of morphine in a rat model of prostate bone metastasis. Pain Med. 2012 Nov;13(11):1444-56. [6]. Michel MC, et al. Unexpected frequent hepatotoxicity of a prescription drug, flupirtine, marketed for about 30 years. Br J Clin Pharmacol. 2012 May;73(5):821-5.
 m.cnreagent.com
m.cnreagent.com