DDR1-IN-2 是盘状结构域受体 1 抑制剂 (IC50:13.1 nM),对 DDR2 的抑制作用相对较弱 (IC50:203 nM)。
产品描述
DDR1-IN-2 (DDR1 inhibitor 7rh) is a potent, selective, ATP-competitive Discoidin domain receptor 1 (DDR1) inhibitor (IC50: 6.8 nM in cell-free kinase assays).
体外活性
DDR1-IN-2 inhibited the enzymatic activity of DDR1, with IC50 values of 6.8 nM, but were significantly less potent in suppressing the kinase activities of DDR2, Bcr-Abl, and c-Kit. It bound with DDR1 with a Kd value of 0.6 nM, while it was significantly less potent to the other 455 kinases tested. It also potently inhibited the proliferation of cancer cells expressing high levels of DDR1 and strongly suppressed cancer cell invasion, adhesion, and tumorigenicity [1]. Pharmacologic inhibition of DDR1 with an ATP-competitive orally available small-molecule kinase inhibitor (DDR1-IN-2) abrogated collagen-induced DDR1 signaling in pancreatic tumor cells and consequently reduced colony formation and migration [2].
体内活性
DDR1-IN-2 possessed good PK profiles, with oral bioavailabilities of 67.4% [1]. The inhibition of DDR1 with DDR1-IN-2 showed striking efficacy in combination with chemotherapy in orthotopic xenografts and autochthonous pancreatic tumors where it significantly reduced DDR1 activation and downstream signaling, reduced primary tumor burden, and improved chemoresponse [2].
激酶实验
The functional assays of compounds on the kinase activities of c-kit and Abl were determined using the FRET-based Z'-Lyte assay system according to the manufacturer's instructions. Tyrosine 2 Peptide was used as Abl substrate and Ser/Thr 6 peptide was used as the substrate for c-kit. The reactions were carried out in 384-well plates in a 10 μl of reaction volume with an appropriate amount of kinases in 50 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM EGTA, and 0.01% Brij-35. The reactions were incubated 1 hour at room temperature in the presence of 2 μM of substrate with 10 μM of ATP (for Abl1 assays) or 300 μM of ATP (kit assay) and in the presence of various concentrations of the compounds. The development reagent was then added for further 2 hours room temperature incubation followed by the addition of stop solution. Fluorescence signal ratio of 445 nm (Coumarin)/520 nm (fluorescin) was examined on EnVision Multilabel Reader. The effects of compounds on the kinases DDR1 and DDR2 were assessed by using a LanthaScreen Eu kinase activity assay technology. Kinase reactions are performed in a 10 μL volume in low-volume 384-well plates. The kinases in reaction buffer consist of 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA, the concentration of Fluorescein-Poly GAT Substrate in the assay is 100 nM, Kinase reactions were initiated with the addition of 100 nM ATP in the presence of serials of dilutions of compounds. The reactions were allowed to proceed for 1 hour at room temperature before a 10 μL preparation of EDTA (20 mM) and Eu-labeled antibody (4 nM) in TR-FRET dilution buffer are added. The final concentration of antibody in the assay well is 2 nM, and the final concentration of EDTA is 10 mM. The plate is allowed to incubate at room temperature for one more hour before the TR-FRET emission ratios of 665 nm/340 nm were acquired on a PerkinElmer EnVision Multilabel Reader [1].
细胞实验
Adherent Cells were plated in 96-well culture plates with a cell density of 3000-4000 cells/well and treated with indicated compounds by adding 100μL medium containing compounds of various concentrations on the second day. After 72-hour's treatment, MTT was added to each well and incubated for additional 4-5 hours, and the absorbance was measured on a microplate reader at 570nm. Cell growth inhibition was evaluated as the ratio of the absorbance of the sample to that of the control. The results are representative of at least 4 independent experiments [1].
动物实验
Compounds 7rh and 7rj were dissolved in mixed solvents (DMSO : EtOH:Cremophor EL : H2O = 2 : 4 : 4 : 90) as clear solution. The final concentrations were 2.5 mg/mL. Sprague Dawley (SD) rats (male, 4 animals per group) weighted 180~220g were injected intravenously or administrated orally at doses of 5 mg/kg (i.v.) or 25mg/kg (p.o.), respectively. After dose administration, 0.3 mL of the orbital blood was taken at 2.0 min, 10.0 min, 30.0 min, 1.0 h, 2.0 h, 3.0 h, 4.0 h, 6.0 h, 8.0 h, 12.0 h, 21.0 h, 24.0 h, 30.0 h, 36.0 h, 48.0 h, and 72.0 h. Samples were stored at -70℃ until shipment to the analytical laboratory and tested by HPLC/MS using propranolol as internal standard to measure the compound concentration in the blood [1].
Cas No.
1429617-90-2
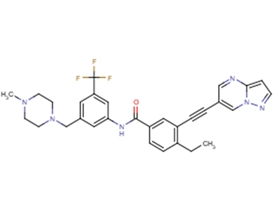
分子式
C30H29F3N6O
分子量
546.59
别名
DDR1 inhibitor 7rh
储存和溶解度
DMSO:12 mg/mL (21.95 mM)
Powder: -20°C for 3 years
In solvent: -80°C for 2 years
 m.cnreagent.com
m.cnreagent.com