| 包装 | 价格(元) |
| 5mg | 电议 |
| 10mg | 电议 |
| 200mg | 电议 |
Kinase experiment: | Inhibition of human cytochrome P450 activities is determined in duplicate in pooled human hepatic microsomal fractions following current scientific and regulatory guidelines. Reaction conditions are linear with respect to incubation time and hepatic microsomal protein concentration. Substrates are present at concentrations equal to or less than their respective Km values determined under the same reaction conditions. Metabolite and/or substrate concentrations are determined using specific, internal standard controlled HPLC MS/MS assays. For reactions monitoring metabolite formation there is less than 20% consumption of substrate during the reaction. Unless otherwise noted microsomal fraction, diluted in potassium phosphate buffer, is preincubated with substrate and inhibitor for 5 min at 37℃ and the reaction initiated by the addition of an NADPH generating system followed by further incubation at 37℃ with shaking. Enzyme-selective positive control inhibitors are tested in parallel. At appropriate times aliquots of the mixture are removed and the reaction terminated by addition to a mixture of methanol and acetonitrile containing the respective internal standard. After centrifugation aliquots of the supernatant are subjected to HPLC-MS/MS analysis. |
Cell experiment: | Five-fold serial dilutions of the tested compounds are prepared in triplicate in 96-well plates. MT-2 cells are added to plates at a density of 20,000/well in a final assay volume of 200 μL. After a 5-day incubation at 37℃, the cytotoxic effect is determined using a cell viability assay. One hundred μL media is removed from each well and replaced with 100 μL of phosphate-buffered saline containing 1.7 mg/mL XTT and 5 μg/mL PMS. Following 1-hour incubation at 37℃, 20 μL of 2% Triton X- 100 is added to each well and absorbance is read at 450 nm with a background subtraction at 650 nm. The data are plotted as cell viability vs. drug concentration. Cell viability is expressed as a percentage of the signal from untreated samples (0% cytotoxicity) after the subtraction of signal from samples treated with 10 μM of Podophyllotoxin (100% cytotoxicity). The CC50 value is calculated from the inhibition plots as the concentration of drug which inhibits cell proliferation by 50%. |
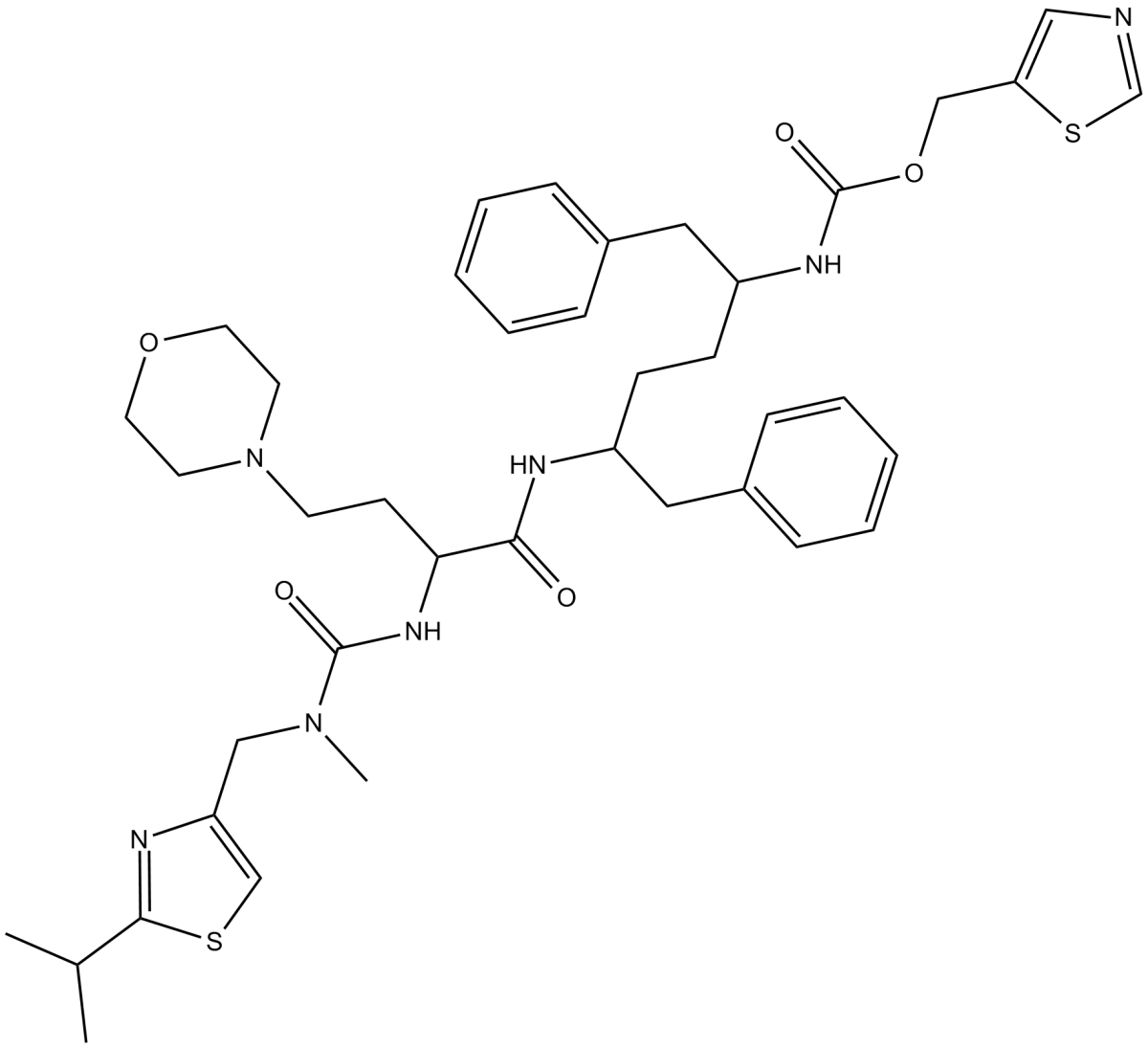
| 产品描述 | Cobicistat is a selective inhibitor of cytochrome P450 (CYP) 3A enzymes with IC50 values ranging from 0.03 to 0.285μM [1]. Cobicistat is a selective CYP3A inhibitor without intrinsic anti-HIV activity and is indicated in EU as a pharmacokinetic enhancer of the HIV-1 protease inhibitors atazanavir and darunavir in adults. Cobicistat is also developed to form a fixed-dose tablet in combination with elvitegravir, emtricitabine and TDF. Cobicistat inhibits CYP3A in vitro across a variety of substrates with IC50 values ranging from 0.03 to 0.285μM. Cobicistat is absorbed rapidly after oral administration in patients infected with HIV-1 with the Cmax of 1.2μg/ml. In the 48-week analysis of the phase III study, once-daily oral cobicistat is generally well tolerated. The tolerability of the cobicistat-boosted atazanavir remains similar longer term, with jaundice, ocular icterus and nausea in a 96-week analysis of the phase III study [1]. References: |
 m.cnreagent.com
m.cnreagent.com